在做好了前期的一些预处理后,我们便可以对数据进行聚类分析去得到一些我们想要的初步分析结果了,对细胞进行聚类主要有两种方法:聚类算法(clustering algorithms)和社区检测算法(community detection methods)。目前单细胞聚类的主流方法是在单细胞数据的knn图上通过社区检测算法(包括louvain和leiden)进行聚类分析。
Clustering
首先利用挑出的高异质性基因进行PCA,进一步降低数据的维数。
1 | # PCA线性降维 |
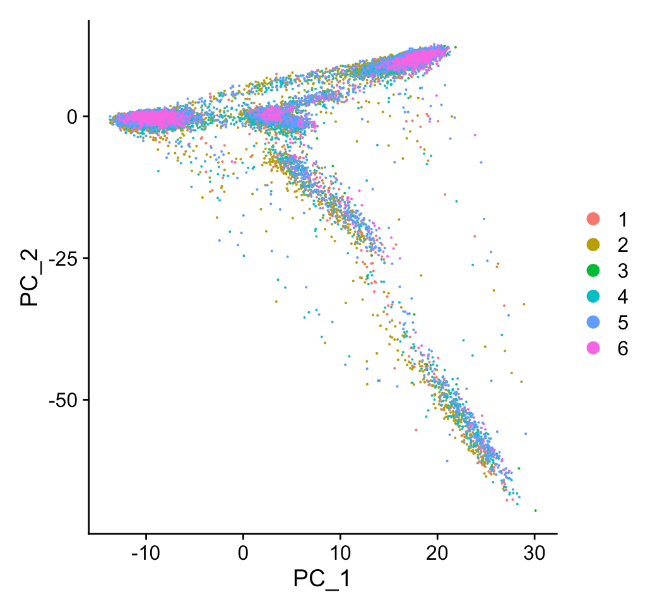
pcaPlot为根据PCA的前两维绘制的散点图,可以大致看出不同的样本的聚集情况。
elbowPlot为碎石图,可以看出每个pc中所包含的信息量,通常用于确定接下来计算邻居结点或者计算Umap时选用多少个主成分。

从碎石图中我们可以看到,主成分所含的信息量在大概pc8-pc10时有一个显著的下降,这个显著下降的点被称为”elbow”, 我们通常选择elbow前的pcs做后续的分析。
对于pcs的选择,Seurat教程给出了一些建议:
- We encourage users to repeat downstream analyses with a different number of PCs (10, 15, or even 50!). As you will observe, the results often do not differ dramatically.
- We advise users to err on the higher side when choosing this parameter. For example, performing downstream analyses with only 5 PCs does significantly and adversely affect results.

确定了用于后续分析的维数后,就可以开始进行聚类了。目前比较通用的方法是先计算出单细胞簇的knn图FindNeighbors(),再在knn图上使用louvain算法检测出细胞的分簇FindClusters(), 最后通过UMAP图可视化分簇RunUmap()。
1 | # 计算邻居 |

也可以使用t-sne图进行可视化
1 | AllCell <- RunTSNE(AllCell, dims = 1:10) |
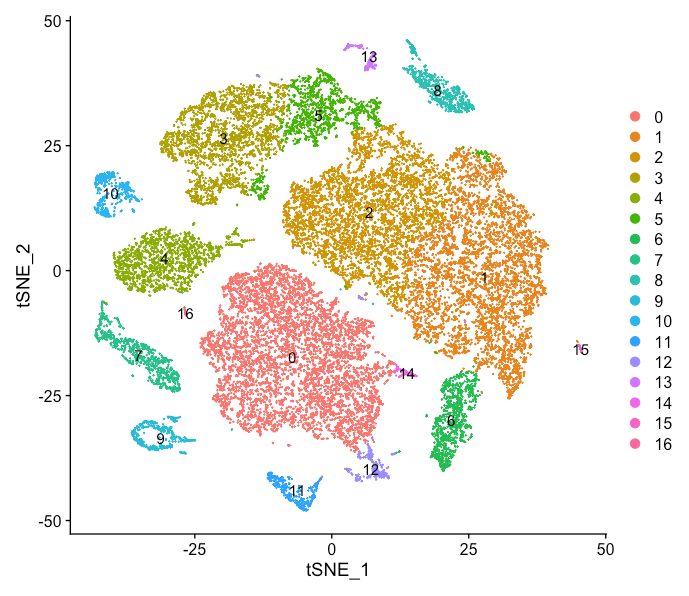
Seurat包中的图都是基于ggplot2绘制的,因此可以通过ggplot2对绘制的图进行设置:
1 | DimPlot(AllCell, reduction = "umap", label = T) +theme( |

1 | DimPlot(AllCell, reduction = "tsne", label = T) +theme( |

SeuratObject的构成
现在我们可以回过头来,看看AllCell里到底有什么
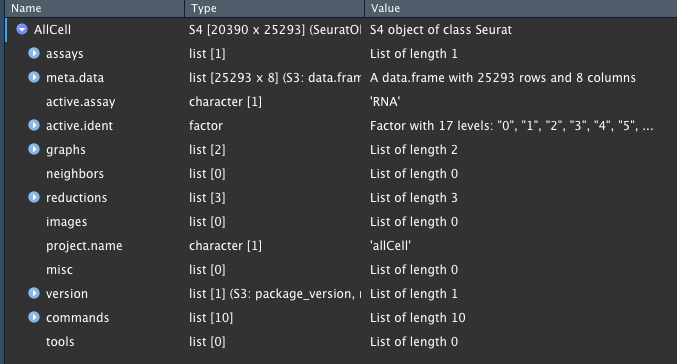
| slot | function |
|---|---|
| assays | A list of assays within this object |
| meta.data | Cell-level meta data |
| active.assay | Name of active, or default, assay |
| active.ident | Identity classes for the current object |
| graphs | A list of nearest neighbor graphs |
| reductions | A list of DimReduc objects |
| project.name | User-defined project name (optional) |
| tools | Empty list. Tool developers can store any internal data from their methods here |
| misc | Empty slot. User can store additional information here |
| version | Seurat version used when creating the objec |
| …… | …… |
挑出几个重要的介绍一下:
Assays
assays中存放着原始的表达矩阵,基因名,barcode等信息,
如果想获得数据中的基因名,可以直接使用
rownames()如
Output: [1] “Xkr4” “Gm1992” “Rp1” “Sox17” “Gm37323” “Mrpl15”
也可以使用稍微复杂的表达方式:
Output: [1] “Xkr4” “Gm1992” “Rp1” “Sox17” “Gm37323” “Mrpl15”
barcode同理,可以使用
colnames()或AllCell@assays[["RNA"]]@data@Dimnames[[2]]获取
AllCell@assays[[“RNA”]]中,在聚类的标准流程走完后通常含有三种数据:counts, data, scale.data.
counts中包含的是原始表达矩阵中的counts值;
data中包含的是Normalization之后的值;
scale.data中包含的是Scale之后的值;
在一些Seurat函数如FindMarkers()中,会有一个参数slot可以选择用于计算的是何种数据形式,默认值为”data”,即使用Normalization之后的数据进行计算。
Meta.data
meta.data 元数据,里面存放着对每个细胞的描述, meta.data中的列可以直接通过AllCell$获取:
在此例中
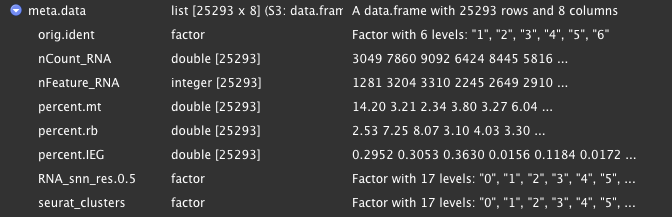
orig.ident为读入样本时对样本的编号,nCount_RNA是每个细胞的总Count数,nFeature_RNA是每个细胞表达的基因数。
percent.mt, percent.rb, percent.IEG是我们计算出的线粒体基因,核糖体基因以及即早基因在每个细胞中所占比例:
1 | AllCell[["percent.mt"]] <- PercentageFeatureSet(AllCell, pattern = "^mt-") # 计算每个细胞中线粒体基因的百分比 |
RNA_snn_res.0.5和Seurat_clusters是每个细胞所属的分簇;
meta.data中可以任意添加新列以描述各个细胞的信息,例如我们从Moffitt的原文得知,前三个样本(1,2,3)是雌鼠。后三个样本(4,5,6)是雄鼠。我们可以在meta.data中添加这一信息:
1 | sex <- fct_collapse(AllCell$orig.ident, "female" = c("1", "2", "3"), "male" = c("4", "5", "6")) |
在Umap图中,我们可以通过group_by参数查看我们关注的信息的分布情况:
1 | DimPlot(AllCell, group.by = "sex", shuffle = T) |

Shffule = T可以按随机顺序绘制点,防止色彩被盖住看不到分布情况。
Active.*
Active.assay和active.ident表示当前默认的数据和当前默认的meta.data,一些函数如FindAllMarker()会根据active.ident计算当前默认分类中的差异表达基因。可以通过Ident()切换,如:

当前默认的active.ident为聚类出的结果。
1 | Idents(AllCell) <- "sex" |
切换成性别为当前默认ident,这样在调用FindAllMarker()函数时就会计算不同性别之间的差异基因。
reduction
reduction中保存了降维计算(pca, umap, tsne)的信息。
主要是图中各个点的坐标:

Commands
Commands保存了之前对数据进行的操作的信息,其中包括该命令运行的时间以及使用的参数等等。

Ref:
Current best practices in single-cell RNA-seq analysis: a tutorial