在这一步我们对加载入Seurat的数据进行进一步的质量控制,剔掉一些质量较差的细胞。其实在读入数据时,我们已经剔除了一部分的基因,例如我们筛掉了表达基因少于200个的细胞和在少于三个细胞中表达的基因。而在这一步,我们会从其它的一些指标来对我们的数据进行质控。
接下来我们对数据进行Normalization,这样做好处多多,可以使数据之间具有可比性,并且可以使数据的分布近似于下游分析的假设(正态分布)。但值得注意的是,理论上没有一种Normalization的方法适用于所有数据,我们应该根据数据集的情况选择合适的Normalization方法。
该流程所用的单细胞数据集来自2018年发表在Science上的Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. 该文章使用10x Genomics对6只小鼠下丘脑的POA(视前区)进行了单细胞转录组测序,测序结果发布在GEO数据库的GSE113576上。
Quality Control(QC)
在QC这一步,我们主要根据三个标准来看细胞测序的质量:nFeature_RNA(细胞表达的基因数), nCount_RNA(细胞表达的总的mRNA分子(UMI)数), percent.mt(细胞中线粒体基因所占的比例)
计算线粒体基因的占比:
1 | AllCell[["percent.mt"]] <- PercentageFeatureSet(AllCell, pattern = "mt-") |
PercentFeatureSet()用于计算细胞中特定的基因(Feature)的占比,线粒体基因都有一个前缀”mt-“,可以使用”mt-“去匹配所有的线粒体基因。
某些数据集中的线粒体基因可能会标为”MT-“,可以先查看一下基因命名确认一下。
绘制小提琴图来查看这三个标准的在数据集中的分布情况:
1 | VlnPlot(AllCell, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size = 0) |

小提琴图横坐标的Identy为每个样本的编号,从这张图我们可以看出每个样本的数据质量。
利用散点图查看不同的标准之间的相关性:
1 | plot1 <- FeatureScatter(AllCell, feature1 = "nCount_RNA", feature2 = "percent.mt") |

FeatureScatter()函数还有很多其它的作用,参数Feature1, Feature2也可以是我们感兴趣的基因,这样我们就可以看到感兴趣的两种基因的共表达情况,当然最好在Normalization等标准流程走完以后再进行共表达的分析。
我们也可以通过颜色添加第三维,将这三个标准展示在同一张图上,但Seurat并没有直接提供函数画出这种图,我们可以ggplot2自己画一下:
1 | testdata <- FetchData(AllCell, vars = c('nCount_RNA', 'nFeature_RNA', 'percent.mt')) |

以上过程为大部分教程中的标准的QC依据,在实际分析中我们可能还会选择一些其它的标准做QC。例如在Moffitt的原文中,作者还考察了核糖体基因(以Rps或Rpl开头)和即早基因(Fos, Egr1, Npas4)的含量,我们也可以考察这两种基因的含量:
1 | AllCell[["percent.rb"]] <- PercentageFeatureSet(AllCell, pattern = "^Rp[ls]") # 计算每个细胞中核糖体基因的百分比 |
即早基因是一组在受到一系列外界刺激后迅速并且短暂激活的基因。
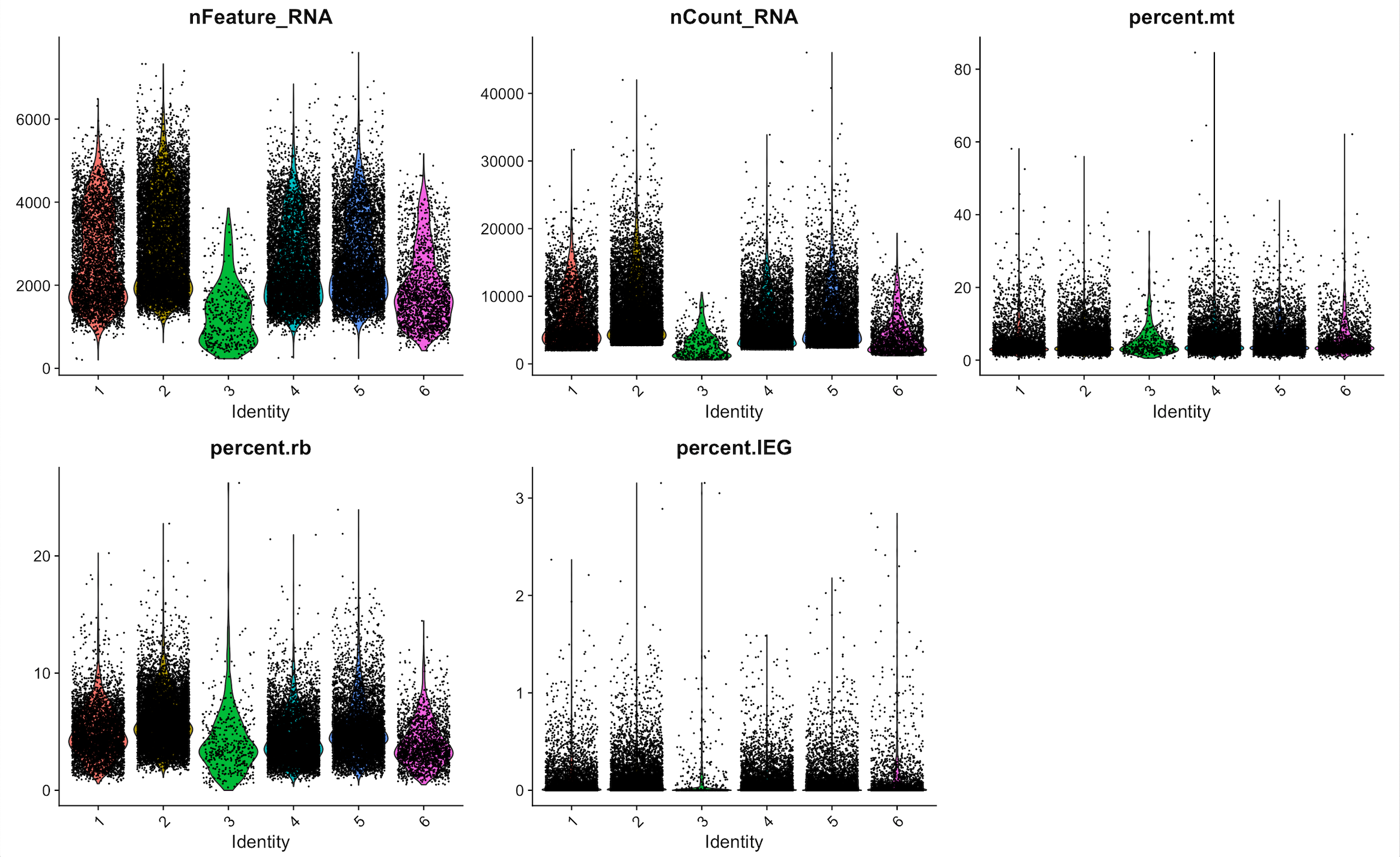
根据绘图的结果,确定筛选的阈值,并进行过滤,第一次对数据集进行分析的时候筛选的标准应当尽量宽松或者不进行筛选。
1 | #其实只是一个取子集的过程 |
Normalization
接下来我们对数据进行Normalization:
1 | AllCell <- NormalizeData(AllCell, normalization.method = "LogNormalize", scale.factor = 10000) |
NormalizationData()函数中的normalization.method中提供的默认参数为“LogNormalize”,该方法是对原始的Counts进行CPM转化后在进行log转化,因此在此步使用”LogNormalize”后不需要对数据进行进一步的对数转换。
在此步一般使用默认参数即可,除了使用Seurat中的
NormalizationData(), Normalization还有许多种方法,例如使用Scran包对数据进行scran(使用卷积的CPM改进版),使用Scone包来评估各个Normalization方法对数据集的效果等等,每种方法的原理和使用范围也不同,在以后会提到。CPM,TPM等Normalization方法是从bulk RNA-seq借鉴过来的,但是用在单细胞转录组这种十分稀疏的数据集可能会出现各种各样的问题。因此会出现各种各样的对Normalization方法,但总的来说,不存在一种方法对所有的数据集都适用,理论上我们应当根据数据集的不同选择不同的Normalization的方法。
放一张xkcd上的图:

寻找高异质性的基因:
1 | AllCell <- FindVariableFeatures(AllCell) |
在这一步我们要选择在细胞之间表达量差异最大的一部分基因,这样接下来的聚类等操作并不会计算所有的基因。而是会根据这些高异质性基因进行聚类。
寻找高异质性基因是一种特征选择(Feature Selection)的过程。
这一步一般来说也只用默认参数即可。
绘制高异质性基因:
1 | # 异质性最高的十个基因 |

对每个细胞的基因表达量进行scale:
1 | # Scaling the data |
至此,原始表达矩阵的转化已经基本完成,接下来可以着手对细胞进行聚类了。
Ref:
PercentageFeatureSet function - RDocumentation
Plotting a 3rd dimension (color by expression) for FeatureScatter #3544
Current best practices in single-cell RNA-seq analysis: a tutorial