进行数据分析首先要获得数据,在这里先介绍一下如何从GEO数据库上下载别人发表的单细胞转录组数据库的表达矩阵,以及如何把数据加载到Seurat中。
该流程所用的单细胞数据集来自2018年发表在Science上的Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. 该文章使用10x Genomics对6只小鼠下丘脑的POA(视前区)进行了单细胞转录组测序,测序结果发布在GEO数据库的GSE113576上。
使用GEOquery下载表达矩阵文件
打开GSE113576的页面,我们所需要的文件在Supplementary file中:

基于10x Genomics测序的数据库,Supplementary file的三个文件通常来自于CellRanger, CellRanger是10X genomics公司为单细胞RNA测序分析量身打造的数据分析软件,可以直接输入Illumina 原始数据(raw base call ,BCL)输出表达定量矩阵,通常会使用CellRanger进行上游处理得到表达矩阵。
- *barcode.tsv.gz中包含测序的细胞barcode等信息;
barcode是单细胞测序中唯一编码一个细胞的碱基序列“身份证”
- *genes.tsv.gz中包含测序的基因名等信息;
CellRanger 2和CellRanger 3该文件的输出结果有所不同。CellRanger 2输出的文件名为*genes.tsv.gz,而在CellRanger 3中,该文件被命名为*features.tsv.gz. 内部表格格式也有所不同。
可以在此直接点击下载,也可以使用GEOquery这个R包进行下载:
1 | library(GEOquery) |
getGEOSuppFiles()用于下载GEO数据库中的Supplementary file文件,baseDir选择文件需要存储的位置。
下载完成会得到三个*.gz的压缩文件。
在R中载入数据
在R中载入相应的包:
1 | library(Seurat) |
Seurat为主要的分析单细胞测序数据的包,tidyverse用于数据的转换与可视化,patchwork用于拼接绘制的图。
使用Read10X()进行读取数据:
1 | raw.data <- Read10X(data.dir = "data/seurat/") |
Read10X()函数只能接受一个文件夹作为输入,这个文件夹中要包含三个文件:barcode.tsv.gz, genes.tsv.gz(Featrues.tsv.gz), matrix.mtx.gz. 因此在读取数据前请删掉下载的数据文件的前缀。
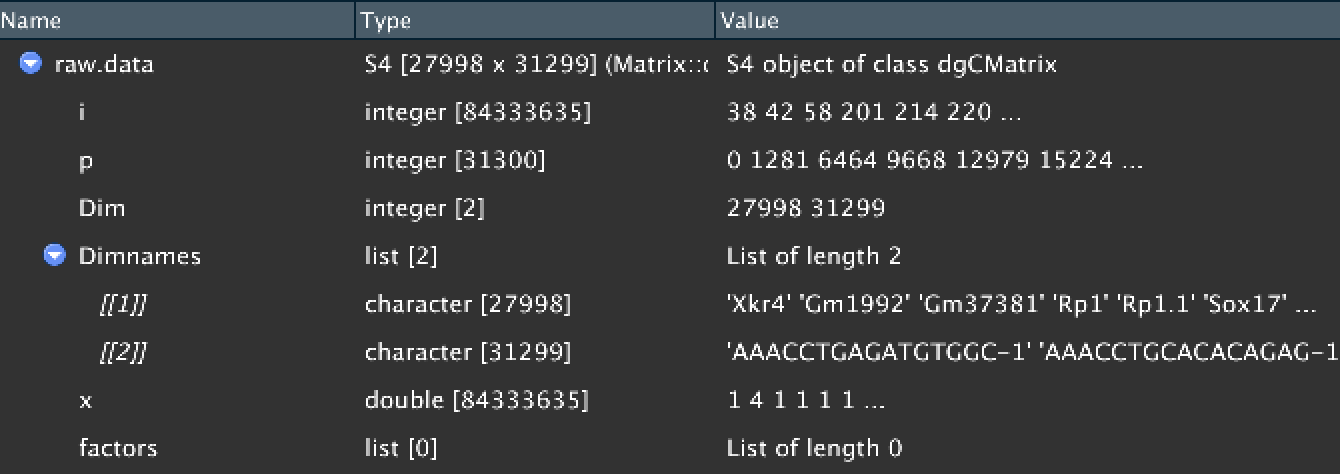
raw.data:
使用CreateSeuratObject()创建Seurat对象,接下来的标准分析流程都以该SeuratObject为基础,分析的信息也会存储在SeuratObj中:
1 | AllCell <- CreateSeuratObject(counts = raw.data, project = "allCell" |
SeuratObj中的信息此处可以先不看,等跑完了聚类分析的基本流程后,再来看Seurat中的各个属性的含义。
CreateSeuratObject()中min.cell = 3表示挑选出至少在三个细胞中表达的基因,min.features = 200表示挑选出至少表达200个基因的细胞,names.delim和names.field用于提取载入的细胞名中的批次等信息。
例如,在’AAACCTGAGATGTGGC-1’细胞中,”-“前一部分是barcode序列,”1”表示样本信息,代表这是第一个样本中的细胞,names.delim = “-“表示barcode序列和样本信息用短横分割([1]-[2])。 names.field = 2表示”-“分割的第二部分([2])为我们想要的样本信息。
至此,单细胞转录组的数据已经被载入到Seurat中,可以对其进行分析了。
Ref.