
单细胞转录组文献分析方法[七]:Interneuron origin and molecular diversity in the human fetal brain


单细胞转录组文献分析方法[六]:Transcriptomic analysis links diverse hypothalamic cell types to fibroblast growth factor 1-induced sustained diabetes remission

单细胞转录组文献分析方法[五]:Spatially organized multicellular immune hubs in human colorectal cancer

单细胞转录组文献分析方法[四]:Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease


单细胞转录组文献分析方法[三]: Single-cell transcriptomic analysis of Alzheimer's disease


单细胞转录组文献分析方法[二]: Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons

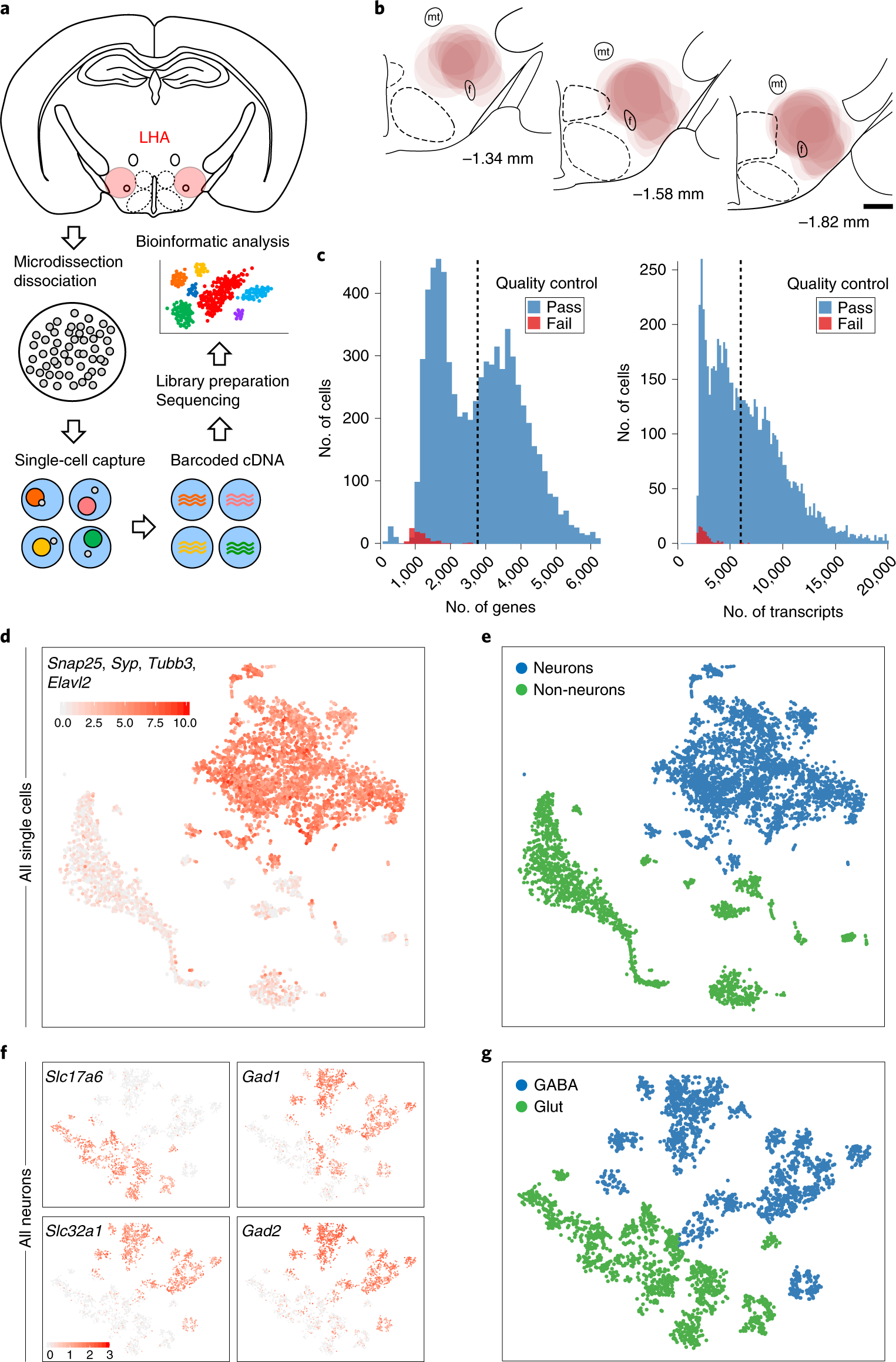
单细胞转录组文献分析方法[一]: Decoding molecular and cellular heterogeneity of mouse nucleus accumbens

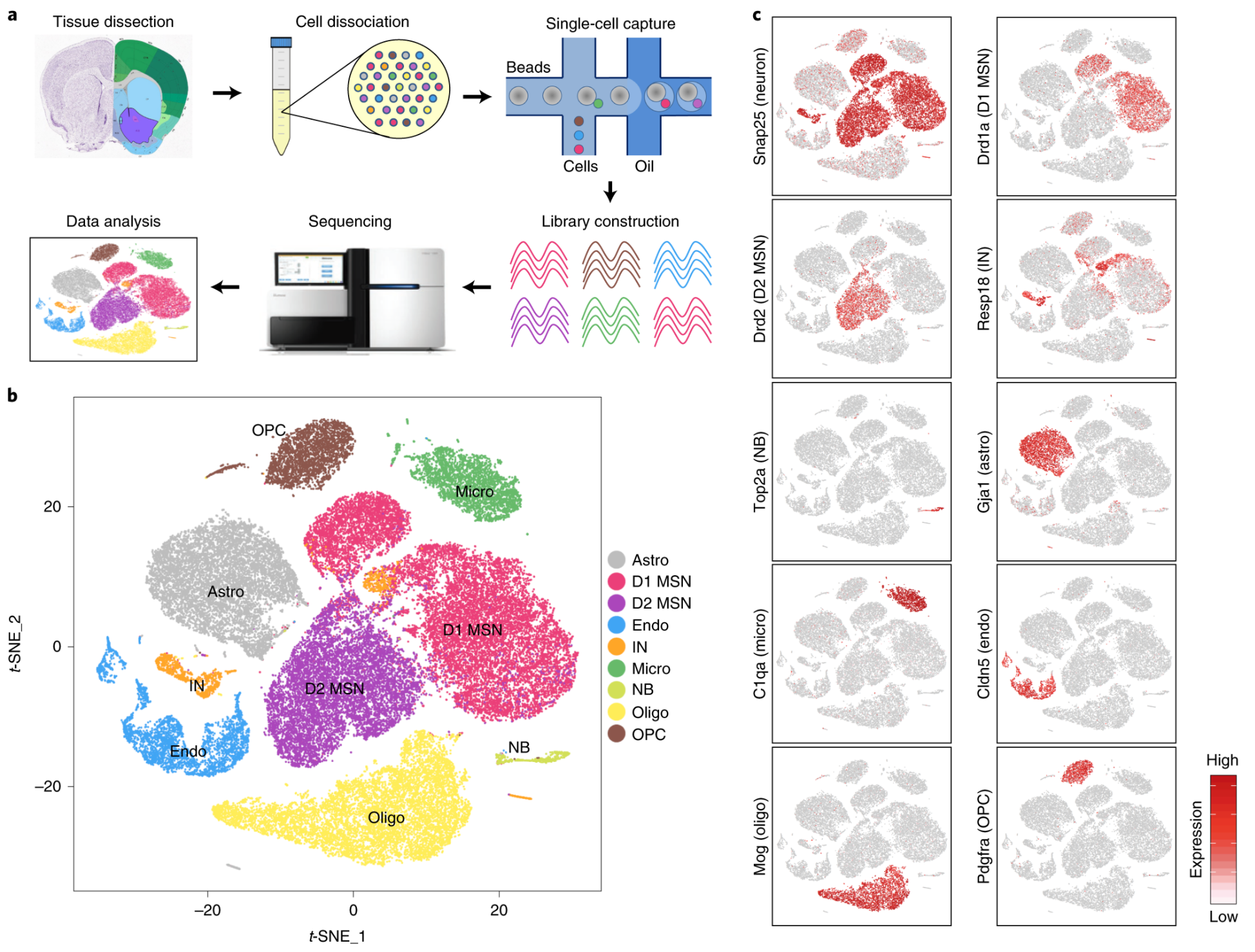
R单细胞数据分析流程(三)Clustering
在做好了前期的一些预处理后,我们便可以对数据进行聚类分析去得到一些我们想要的初步分析结果了,对细胞进行聚类主要有两种方法:聚类算法(clustering algorithms)和社区检测算法(community detection methods)。目前单细胞聚类的主流方法是在单细胞数据的knn图上通过社区检测算法(包括louvain和leiden)进行聚类分析。
R单细胞数据分析流程(二)QC amd Normalization
在这一步我们对加载入Seurat的数据进行进一步的质量控制,剔掉一些质量较差的细胞。其实在读入数据时,我们已经剔除了一部分的基因,例如我们筛掉了表达基因少于200个的细胞和在少于三个细胞中表达的基因。而在这一步,我们会从其它的一些指标来对我们的数据进行质控。
接下来我们对数据进行Normalization,这样做好处多多,可以使数据之间具有可比性,并且可以使数据的分布近似于下游分析的假设(正态分布)。但值得注意的是,理论上没有一种Normalization的方法适用于所有数据,我们应该根据数据集的情况选择合适的Normalization方法。